CMMB 461 – Microarray Analysis Assignment 2
Hello, dear friend, you can consult us at any time if you have any questions, add WeChat: daixieit
CMMB 461 – Microarray Analysis Assignment 2
Instructions
Type your answers (including figures) in the provided question sheet document:
CMMB 461 Assignment 2 - Questions
● There is no page limit for this assignment, but your responses should be concise and clearly answer each question. No more than 2-3 sentences max should be needed to answer any question.
● Do NOT submit this Instruction sheet with your assignment.
● The work that you submit must be your own original work in your own words. University of Calgary plagiarism regulations apply.
Save your question sheet as a PDF file with the format name:
Last Name_First Name_Assignment_2_Questions
Save your R code once complete with the format name:
Last Name_First Name_Assignment_2_R
If you have any questions about this assignment, contact Kurtis Marno Jones – [email protected]
You will need to upload 2 files total to the Assignment D2L Dropbox when submitting as follows:
1. Your completed question sheet saved as a PDF as described above
2. Your completed R code saved with the name listed above
All assignment components must be uploaded to D2L (digital) Dropbox prior to 11:59 PM on Friday November 5th.
! Make sure you leave yourself enough time to complete this assignment. Getting everything working and running properly can take several hours.
Materials
Download the following files from the Assignment 2 folder on D2L
Raw Data Files:
gene.a.del.spot (raw microarray data of empty vector vs the gene A deletion)gene.a.OE.spot(raw microarray data of empty vector vs the gene A overexpression)gene.b.del.spot (raw microarray data of empty vector vs the gene B deletion strain)gene.b.OE.spot(raw microarray data of empty vector vs the gene B overexpression)
pombe.galPombeExperement.txt
R Script File:
CMMB461_Assignment_2.R
MEME Files:
GENE_B_MEME_FILE.txt
Programs needed to be downloaded
RR StudioJavaJava TreeView
Background information:
The main objectives of the assignment are to reinforce certain concepts in lectures and to gain practical learning in the analysis of expression microarray (transcriptome) data. The concepts include microarray data normalization, clustering and generation of a heat map with dendogram, functional enrichment analysis of differentially regulated genes between a mutant strain and wild type and the use of a genome browser (PomBase). You will utilize the provided microarray data to investigate two transcription factors, determine their function and activity, as well as identify their target genes.
Transcription factors (TFs) are proteins that turn on or off specific target genes by regulating the level of mRNA synthesis. They function by binding to a DNA motif, which is a common short sequence (5-20 nucleotides long), located in the promoter or enhancer of their target genes.
Microarray expression profiling can be used to identify the target genes of transcription factors. This can be accomplished by comparing two sets of data, first by comparing the transcriptome of a wild-type strain (the transcription factor is present) to a transcription factor deletion mutant (loss-of-function / LOF allele) in which the transcription factor is not present because the gene has been deleted. The target genes of the transcription factor should be differentially regulated when comparing the transcriptomes of these two strains. If the transcription factor activates the transcription of its target genes, then the relative abundance of the mRNA of the target genes would be lower in the deletion strain compared to wild type.
However, if the transcription factor represses the transcription of its target genes, then the relative abundance of the mRNA of the target genes would be higher in the deletion strain compared to the wild type.
The second set of data looks at the comparison between the transcriptome of an empty vector control strain (the transcription factor is present at normal activity) to a hyperactivation of the transcription factor also known as an overexpression (gain-of-function / GOF) strain. The transcription factor gene is overexpressed by cloning the open reading frame into a vector with a strong promotor. The empty vector control strain contains the same overexpression vector, except there is no transcription factor gene cloned into it (hence the term “empty vector”). In this case, we would expect that if the transcription factor is an activator, then the mRNA abundance of the target genes would be higher in the overexpression strain relative to the empty vector control strain. In contrast, if the transcription factor is a repressor, then the mRNA abundance of the target genes would be lower in the over expression compared to the empty vector control strain.
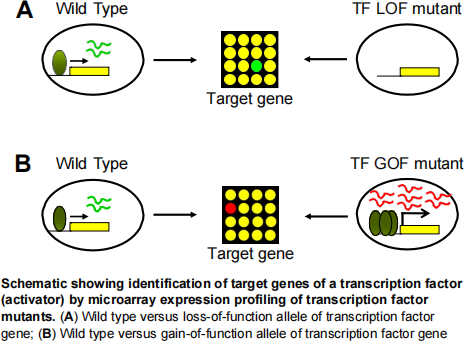
The data provided in this assignment are derived from microarray expression profiling of loss- of-function and gain-of-function alleles of three fission yeast (Schizosaccharomyces pombe) genes (Genes “A” and “B”) that encode for transcription factors A and B, respectively. Genes A and B encode either a transcriptional activator or a transcriptional repressor (you will have to figure out which one). Microarray expression profiling was performed on a: (1) gain-of-function overexpression (OE) allele of gene A or gene B versus an empty vector control strain; (2) loss-of-function null allele (del) of gene A or gene B versus the wild-type control strain. It is expected that both genes A and B would show highly differential gene expression (log2 ratios) in the mutant alleles relative to the control strains in these microarray experiments. Your aim is to determine the identity of the transcription factors A and B, their target genes, and biological function from the microarray data.
Procedure
1. Program Installation
1.1 Download and install R studio.
● The software we are going to be using to do the initial preprocessing and clustering of microarray data is R studio.
● First you need to download and install R which is the base code for R studio. Follow the link below and select the version of R for your system.
https://cran.rstudio.com/index.html
● Once R is installed, we can then install R studio. Follow the link below to download and install R studio, choose the version of R Studio that is applicable for your operating system.
https://www.rstudio.com/products/rstudio/download/#download
1.2 Download and install Java TreeView.
● To generate a heatmap and dendrograms of your clustered microarray data, you will use TreeView.
● TreeView is written in Java so first install Java by following the link bellow and following the instructions to install it.
https://java.com/en/
! NOTE: If you use a Mac, you will ALSO need to install the legacy Java Runtime 6 from Apple. You can download it from the link below.
https://support.apple.com/kb/DL1572
● Once Java is installed you can go ahead and download TreeView from the link below.
http://sourceforge.net/projects/jtreeview/files/jtreeview/1.1.6r4/
● On Windows, extract this folder and run the setup application. On Macs, all you need to do is extract it (if your Mac hasn’t already), right-click ‘Java TreeView’, and press Open.
2. Download Assignment Files from D2L
If you have not done so already, download the assignment files (listed above) from the Assignment 2 folder on D2l and make sure that they are all in a single folder on your computer.
3. Microarray Preprocessing and Clustering
Open the CMMB461_Assignment_2.R file (also known as an R Script) using R studio and follow the instructions.
4. Generation of Heat Map and Dendrogram by TreeView
Now it is time to look at the microarray data we just clustered in TreeView. TreeView will make a heatmap and dendrograms of the clustered microarray data. This makes it easier to identify trends (clusters of co- expressed genes) within our microarray data.
4.1 Make sure you have the following set of files before you proceed.
● gene-expression-clustered.atr
● gene-expression-clustered.cdt
● gene-expression-clustered.gtr
4.2 Open TreeView and from the File menu, open the cdt file that R would have saved to the folder you designated as your working directory.
● TreeView should now look something like below with two dendrograms (vertical and horizontal) and a heatmap.
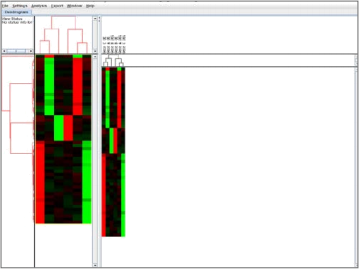
4.3 Next, we look at specific parts of the heatmap.
● We can select different clusters of genes by clicking different branches of the gene dendrogram on the left-hand side of the heatmap.
● By clicking on different parts of the dendrogram you will notice that a zoomed in view of it will show in the panel on the right alongside the corresponding names of the genes in the cluster.
5. Searching for GOTerm enrichment of clustered genes
Now we are going to look at the functional enrichment of certain clusters of genes that we will export from TreeView. We will use a tool called the Princeton GOTerm Finder that uses databases of GO-term (gene ontology) annotations of known or predicted process or function that a specific gene may have. Based on a list of genes that is input, Princeton GOTerm Finder searches for enrichment of a particular function within that list compared to the average chance that a gene within a genome would have the same function.
5.1 Looking at your heatmap, find the most reciprocally regulated cluster of genes between the two microarray experiments. This means the cluster(s) with the greatest amount of green and/or red on the heatmap of the two microarray experiments.
● Which gene cluster should you pick? That requires a bit of background on what genes A and B that we have either overexpressed or deleted are predicted to do. These initial predictions are based off prior studies and databases (such as PomBase).
● The two genes (A and B) encode for either a positive (activator) or negative (repressor) transcriptional regulator. Therefore, the mRNA abundance of the target genes of these two types of transcription regulators are expected to be:
○ up-regulated (red) when the transcriptional repressor is deleted relative to wild-type, or when the transcriptional activator is overexpressed relative to the empty vector control.
○ down-regulated (green) when the transcriptional repressor is overexpressed relative to the empty vector control or when the transcriptional activator is deleted relative to wild-type.
5.2 We will start by selecting the most deferentially regulated clusters for the two gene A microarray experiments (e.g., AOE and Adel columns in the heatmap).
● Select the gene A cluster by clicking on the dendrogram that corresponds to this cluster on the left-hand pane in TreeView.
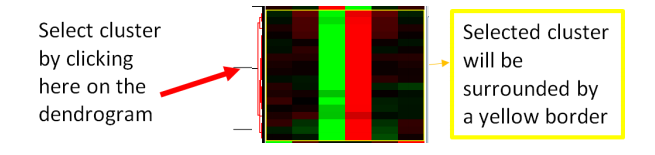
● This should select a cluster of 8 genes.
● Once you have selected this cluster, click the ‘Export’ menu, and choose ‘Save List’.
● Change the name so it ends with ‘_gene A ID’ and click Save.
● This will produce a short text document in your Assignment Files folder.
5.3 We will repeat the same steps for gene B that does not have a function related to gene A. Here, we are not interested in the gene expression of the gene A columns in this case, but the two gene B columns in the heatmap.
● Select the gene B cluster by clicking on the dendrogram that corresponds to this cluster on the left-hand pane in TreeView.

● This should select a cluster of 8 genes.
● Once you have selected this cluster, click the ‘Export’ menu, and choose ‘Save List’.
● Change the name so it ends with ‘_gene B ID’ and click Save.
● This will produce a short text document in your Assignment Files folder.
5.4 Now we will use the Princeton GOTerm Finder:
● Go to this link.
http://go.princeton.edu/cgi-bin/GOTermFinder
○ Where it says, “Upload a file containing lists of genes” click choose file and select the list file ending with “_gene A ID”.
○ Then change the annotation database to “PomBase – S. pombe”.
○ Click ‘Search for GO Terms’ - This may take several minutes to process.
● Go to this link.
http://go.princeton.edu/cgi-bin/GOTermFinder
○ Where it says, “Upload a file containing lists of genes” click choose file and select the list file ending with “_gene B ID”.
○ Then change the annotation database to “PomBase – S. pombe”.
○ Click ‘Search for GO Terms’ - This may take several minutes to process.
6. Searching for putative transcription factor binding sites (DNA motifs)
Now that we have identified two unique sets of possible target genes for transcription factors A and B, we are going to use these lists to identify the putative binding sequence for transcription factor B only. Transcription factor binding sites or DNA motifs exist as similar small sequences (5-20 bp) located in the promoters of their target genes (see figure below).
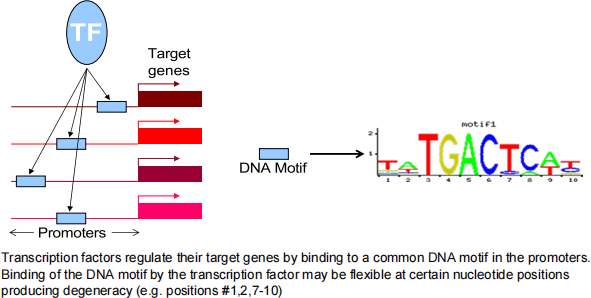
We are going to use a DNA motif-finding tool called MEME and input promoter sequences from the target genes of transcription factor B that wee will manually retrieve from PomBase. MEME searches the input promoter regions of target genes for statistically overrepresented sequences that could potentially be DNA binding sites of the transcription regulator.
6.1 Retrieving Sequences from PomBase:
● Open the list file named ‘gene B_SEQUENCES_MEME’.
● This contains the promotor sequences of 6 of the 8 target genes of transcription factor B that you will need for analysis by MEME. You will need to retrieve the promotor sequence of the remaining two target genes of transcription factor B from PomBase and add it in the correct format to the ‘gene B_SEQUENCES_MEME’ file.
○ fio1
○ fip1
● Go to PomBase.
https://www.PomBase.org/
● Copy the first gene from the above, paste it into the ‘search …’ box, press enter.
● Scroll down to the ‘Sequence’ section.
● Change ‘Include 0 bases upstream’ to 1000 and select 5` UTR.
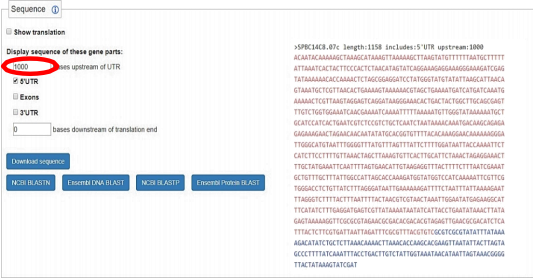
● Select and copy the entire Sequence on the right-hand side.
● Paste this sequence into the ‘gene B_SEQUENCES_MEME’ file.
● Repeat this for the remaining gene and save the file.
6.2 When you have populated your ‘gene B_SEQUENCES MEME’ file with the promotor sequences of the fio1 and fip1 genes upload the file to MEME (below link).
http://meme-suite.org/tools/meme
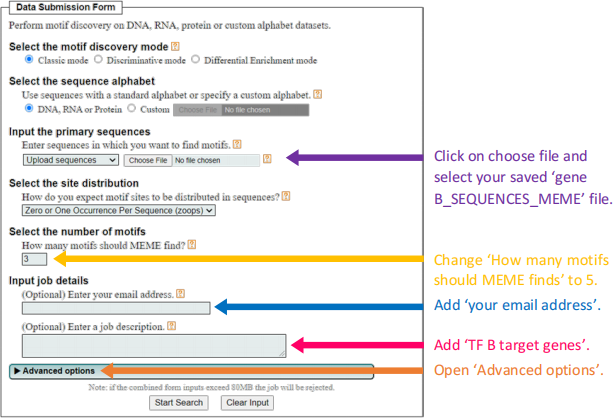
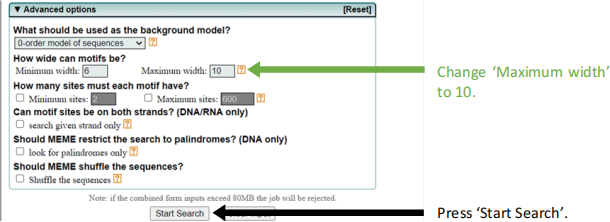
● When your MEME Search finishes, choose ‘MEME HTML output’.

6.3 GOMo (Gene Ontology for Motifs) related terms
● GOMo is an algorithm contained in the MEME tool suite that searches for a gene ontology (GO) category/category that are statistically enriched in a set of target genes that contain a common DNA motif identified in MEME.
● In the ‘Discovered Motifs’ section, click the right arrow for the top DNA motif represented with the most sites in the promotors of the target genes and the highest width look at this
! NOTE: There may be multiple terms that are tied for the most sites and highest width, however only one will be enriched.
● Choose ‘GOMo’ from the ‘Submit to program’ list and press ‘Submit’.
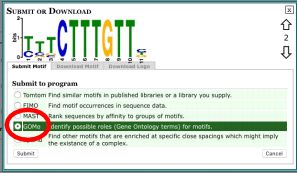
● In GOMo fill out the following:
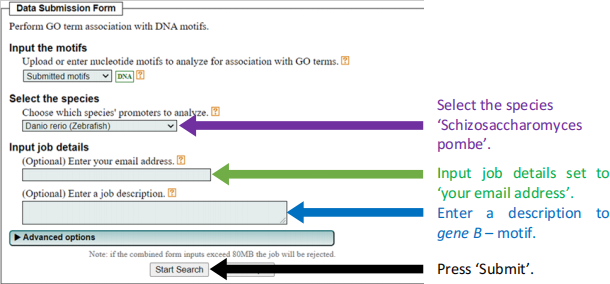
2021-11-09