CHEM0028: CONCEPTS IN COMPUTATIONAL CHEMISTRY Coursework 2022
Hello, dear friend, you can consult us at any time if you have any questions, add WeChat: daixieit
B.Sc. DEGREE 2022
M.Sci. DEGREE 2022
CHEM0028: CONCEPTS IN COMPUTATIONAL CHEMISTRY Coursework
SECTION A
This question relates only to the use of molecular mechanics forcefields and techniques covered by Prof Lewis. It is not necessary to know further details of the protein and inhibitors beyond what is explicitly stated here. There are no marks for identifying the protein further or the source of the material.
Small molecules are used as inhibitors of protein action – as drugs. They most often do this by blocking the active site within the protein. Potential drugs can be screened computationally to determine if they are strongly bound to the protein.
Figure 1 shows a possible conformation of a candidate drug molecule, 4-bromo-2- carboxymethylamide-pyrrole (abbreviation: BCMAP) at the active site of a protein (abbreviation: PR). Figure 2 shows the full protein structure whilst figure 3 shows a known inhibitor of the protein at the site, overlayed with another calculated conformer of BCMAP.
(a) Explain what types of interactions, both intermolecular and intramolecular, that a molecular mechanics forcefield must be able to describe in order to be able to accurately determine the geometry of BCMAP in the protein. Identify which interactions will be the most important to describe accurately. [5 marks – no more than ~1/3 page]
(b) Outline how you would identify likely geometries of BCMAP in the binding site of PR. You need to consider different conformers of BCMAP but only the single active site in PR. Explain how you would rank the more probable geometries for comparison to experimental data on known inhibitors (such as in Figure 3). You should comment on any approximations to the protein structure that may be necessary or reasonable to make. [15 marks – no more than one page]
(c) Outline how you would investigate whether BCMAP would be an effective inhibitor for the protein in competition with the molecules that the protein normally targets, and any solvent. [8 marks]
(d) If the portions of the protein (highlighted in Figure 3) linking the helical structures in PR were to be chemically cleaved, how would you determine computationally the impact of this change in protein structure on the binding of BCMAP in the active site? [7 marks]
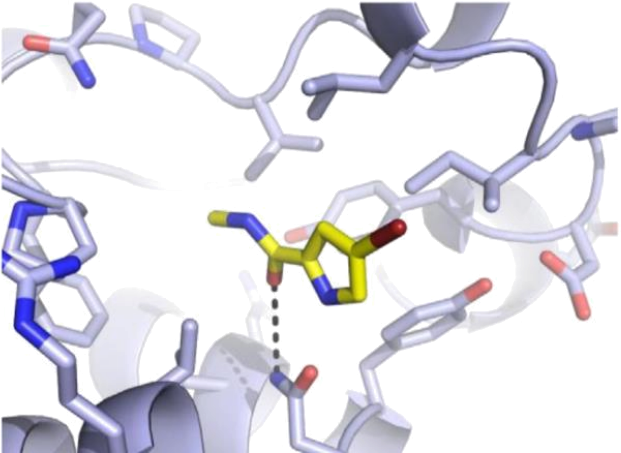
Figure 1. 4-bromo-2-carboxymethylamide-pyrrole (BCMAP) (C, N, O, and Br atoms in yellow, blue, red,and brown, respectively) in the active site of the protein structure.
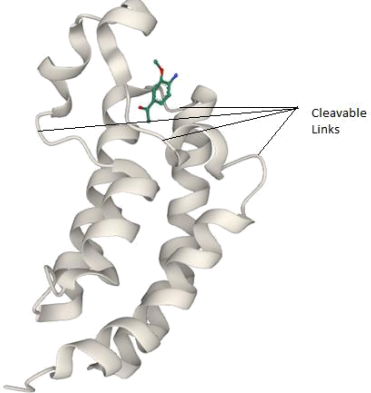
Figure 2. Cartoon (ribbon) structure of the entire protein with a known inhibitor in position, as determined experimentally. Cations and water molecules in the protein structure are omitted for clarity. The links labelled can be cleaved chemically.

Figure 3. An overlay of a known inhibitor (from Figure 2) and an example conformer of BCMAP. Dashed lines indicate hydrogens bonds and electrostatic interactions. The isolated cyan atoms are sodium cations and the isolated red atoms the determined positions of the oxygen in a water molecule.
![]() SECTION B
SECTION B
This question relates to the material from Dr Heleon Quantum Mechanical methods
a) Which of the following chemical species could have their electronic ground state energy and wavefunction satisfactorily described by a restricted Hartree-Fock (RHF) calculation? Explain your reasoning. For those which cannot be described by RHF, suggest one suitable alternative method. [5 marks]
|
i. |
He |
|
ii. |
He+ |
|
iii. |
The allyl anion |
|
iv. |
The allyl radical |
|
v. |
The allyl cation |

b) Critique the following computational schemes. For each, suggest advantages and disadvantages of the proposed calculation and, as appropriate, suggest a more suitable, feasible or accurate alternative. [5 x 5marks]
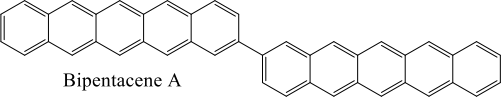
i. Structural optimization of abipentacene A molecule using a complete active- space self-consistent field (CASSCF) calculation in acc-pVDZ basis.
ii. Using Hückel theory to calculate the energy gap between the first excited singlet (s1 ) and first excited triplet (T1 ) states of benzene.
iii. A researcher is computing bond lengths using Hartree-Fock theory. They find that a smaller basis set leads to a result in better agreement with experiment than a larger basis set, and therefore conclude that a smaller basis set is more accurate.
iv. A researcher is calculating the binding energy of an ammonia dimer. They compute the energy of the dimer using DFT ina 6-31G* basis set and subtract from this twice the energy of an isolated ammonia molecule, also using DFT in a 6-31G* basis set.
v. Estimating the first ionization energy of water as minus the energy of the HOMO of a neutral water molecule.
SECTION C
This question relates to the material from Dr Suter on Monte Carlo and multiscale methods
a) What does scale refer to in the context of multiscale modelling? [2 marks]
b) QM/MM and All-Atom/Coarse-Grained Molecular Dynamics are two of the most common multiscale approaches in computational chemistry. Describe the flow of information between the two single-scale models in each of the two methods (use diagrams if appropriate). Explain, with reference to these multiscale models, what is meant by hierarchical and concurrent coupling and give an example of where each approach is most appropriate. [6 marks]
c) Monte Carlo has no inherent timescale. Explain why this is the case. Describe the connection between Metropolis Monte-Carlo and equilibrium properties. [7 marks]
d) Figure 4 shows the chemical structure of the Trp-cage protein. This protein contains 20 amino acid and is one of the fastest folding proteins, with a folding timescale of ≈4 μs. You wish to compute the different possible folded conformations of the Trp-cage protein.
i. Describe a procedure to compute the folded structures of the protein using coarse-grained molecular dynamics in a feasible wall-clock timeframe. [6 marks]
ii. Explain what assumptions you make in the above procedure, and what limitations they could impose on your results. [5 marks]
iii. Explain when Metropolis Monte Carlo methods may be a more appropriate choice than Molecular Dynamics for computing the folded structures of Trp-cage protein. Explain the different properties you can obtain using both methods. [4 marks]
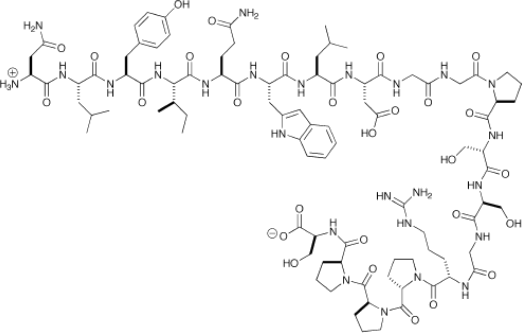
Figure 4: Atomistic structure of the Trp-cage protein.
2023-12-27