COMPSCI 369 Computational Biology FIRST SEMESTER, 2022
Hello, dear friend, you can consult us at any time if you have any questions, add WeChat: daixieit
COMPSCI 369
FIRST SEMESTER, 2022
COMPUTER SCIENCE
Computational Biology
Section A: Computational Biology and Numerical Integration
1. Describe two different ways that computational methods have expanded our ability to study the prisoner’s dilemma. Each description should be at least 2–3 sentences long and should explain why computational methods were necessary, that is, why the approach would have beendifficult or impossible without computers. [8 marks]
2. Answer each of the following questions about Euler integration.
(a) Explain what Euler integration is used for. Include in your explanation an example of a situation where you might use Euler integration. [4 marks]
(b) What is the main assumption that Euler integration depends upon and how does this assump- tion lead to inaccuracies? [4 marks]
(c) Identify a situation where Euler integration would be perfectly accurate and explain why this is the case. [4 marks]
(d) Describe how Euler integration is accomplished (you can use either pseudo-code or mathe- matical notation). Be sure to be clear about what any terms or symbols mean in your expla- nation. [6 marks]
(e) What is the Runge–Kutta (4th order) method and how does it relate to Euler integration? [4 marks]
Section B: Sequence Alignment
3. The partially completed F matrix for calculating the local alignment of the sequences GCT and TAACT is given below. The score matrix is given by s(a, b) = -1 when a ![]() bands(a, a) = 6 while the gap penalty is d = -3.
bands(a, a) = 6 while the gap penalty is d = -3.
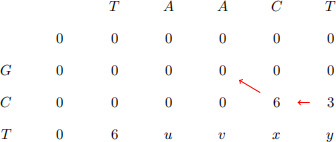
(a) Complete the matrix by finding values for u,v, x and y and showing traceback pointers [4 marks]
(b) Give the score for the best local alignment of these two sequences and provide an alignment that has this score. [3 marks]
4. What is the computational complexity (for time and for memory) for the local alignment algorithm with a linear gap penalty? Justify your answer. [4 marks]
5. If we only wish to find the score of the best local alignment, describe how the local alignment algorithm can be modified to make it more efficient and state the algorithmic complexity (for time and memory) of the resulting algorithm. [6 marks]
6. Consider the Feng-Doolittle progressive alignment algorithm.
|
(a) Describe the role of the X character and why it is important. |
[4 marks] |
|
(b) Explain what iterative refinement is and why it is important. |
[4 marks] |
Section C: Simulation, HMMs and Trees
7. (a) What is the distribution of the random variable that the randomVar pseudo-code below generates given that randomUniform produces a uniformly distributed random variable between 0 and 1? Explain your answer.
var = 0
x = 0
while x < a
if randomUniform() < b
var = var + 1
x = x + 1
return var
[4 marks]
(b) Given methods randunif, sample, randexpand randpoiss, sketch a pseudocode method mutate(A,L,mu,t) that takes a sequence A of length L and mutates it according to the Jukes-Cantor model from time s to time s + t with mutation parameter mu. Allow mutations from a base to itself. [6 marks]
8. Consider an HMM with states A, B, C each of which emit symbols Q, R, S, T. The transitions are given by the following table which has omitted the transition probablities for each state to itself.

The model starts in state A 50% of the time and state B the rest of the time.
The emission probabilities for the model are given by the following table.

(a) Write down the values of the missing elements in the transition matrix. [2 marks]
(b) Sketch a diagram of the HMM, showing all states, possible transitions and transition proba- bilities. Include the begin state but no end state. [3 marks]
(c) How long is the average run of Cs in a state sequence and what distribution, with what parameters, do such runs follow? [3 marks]
(d) What is the joint probability P (x, π) of the state sequence π = AABC and the symbol se- quence x = QTRR? Leave your answer as a product or sum of numbers. [3 marks]
(e) The forward algorithm calculates the quantity P (x) by iteratively calculating the quantities fA (i), fB (i), and fC (i). For the symbol sequence x = QT RR, what probability does fB (4) represent? [2 marks]
(f) For π = AABC and x =QTRR, is P (x) greater than, less than, or equal to P (x, π)? Justify your answer. [2 marks]
(g) Complete the entries i,j and kin the forward matrix below. Remember to show your working. ![]() 0 Q T
0 Q T
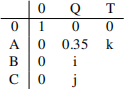
[5 marks]
9. Let the symmetric matrix
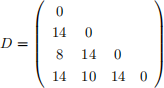
specify the pairwise distances, Dij , between the four sequences x1 , . . . , x4 .
(a) Construct a UPGMA tree from D showing your working. [4 marks]
(b) Will UPGMA or neighbour-joining (or both or neither) reconstruct the correct tree in this case? Explain your answer. [4 marks]
(c) Name an advantage and a disadvantage of neighbour-joining over UPGMA. [2 marks]
10. Consider the four aligned sequences, W,X,Y, and Z:
12345
W: ACCGC
X: ATCAA
Y: ATCAA
Z: AAAGC
(a) Explain what parsimony informative means, and identify the parsimony informative sites in the alignment. [3 marks]
(b) By calculating the parsimony score for each possible tree topology for these four taxa, find the maximum parsimony tree. [4 marks]
(c) If the parsimony model is correct, how many mutations occurred since the most common recent ancestor of these sequences? [2 marks]
(d) State what the main assumptions of parsimony are and under what circumstances they might be reasonable for real sampled sequences. [3 marks]
11. (a) What are the advantages of using a maximum likelihood approach over a parsimony or dis-tance based method to reconstruct trees? [5 marks]
(b) How are tree branch lengths found in a likelihood framework and in a parsimony framework? [4 marks]
(c) How is an acceptable tree found in a likelihood context and how does this differ from how a tree is found in the parsimony context? [4 marks]
2023-06-20