COMPSCI 369 Computational Biology FIRST SEMESTER, 2021
Hello, dear friend, you can consult us at any time if you have any questions, add WeChat: daixieit
COMPSCI 369
FIRST SEMESTER, 2021
COMPUTER SCIENCE
Computational Biology
Section B: Computational Biology and Numerical Integration
1. Your research assistant generated the bifurcation plot below but excluded some important details. Aside from the plot you know that:
i) The difference equation of the system being studied is: xt+1 = exp( ·axt(2)) + b
ii) In this plot, the parameter a = 4.9
iii) When b = 0, there is a periodic attractor.
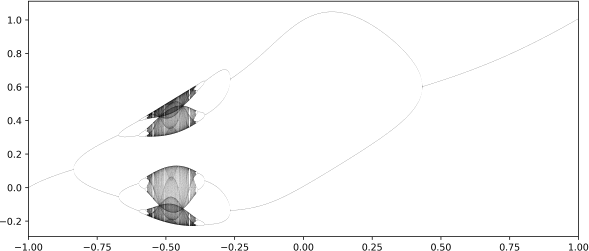
Based on the plot and the information you have, answer the following questions.
(a) What would be an appropriate label for the horizontal axis? [1 mark]
(b) What would be an appropriate label for the vertical axis? [1 mark]
(c) Why is it impossible for someone who only has access to this plot to identify an exact set of parameter values where the system’s dynamics are certain to be chaotic? [2 marks]
(d) Given the information provided above, describe an approximate region in parameter space where this system has a periodic attractor that involves 4 different states. [1 mark]
(e) Describe the technique presented in lectures for creating a diagram like this one. You may use pseudo-code to do so, but are not required to. [10 marks]
(f) The technique you have just described has two important parameters that must be tuned to make the bifurcation diagram legible and accurate. Identify these parameters and explain why the valueselected for each should not be too high nor too low. [7 marks]
Section C: Sequence Alignments and Assembly
2. You are given a function global(A, B, match, mismatch, gap) that solves the global
alignment problem. It returns the optimal score and the optimal alignment when given match/mismatch scores, a linear gap penalty, and a pair of sequences.
(a) Can you use the global function to align two sequences in such a way that the alignment has only matches and gaps? If so explain how, otherwise suggest a modification of global(A, B, match, mismatch, gap) that would enable this. In the latter case, you do not have to use pseudo-code; just explain your idea in one sentence. [2 mark]
(b) You want to calculate the optimal overlap alignment of two sequences of equal length. Can this problem be solved using the global function, while not modifying global? Explain your answer. You may use pseudo-code to do so, but are not required to. [6 marks]
3. The global alignment function global(A, B, match=2, mismatch=-3, gap =-1) pro- ducedthe optimal path traced here:

Answer the following questions and explain your answers.
(a) What will the optimal alignment look like? [2marks]
(b) Is it possible with the given information to calculate the optimal score? If so, write down the optimal score. If not, explain why not. [3 marks]
4. You are given the following profile matrix:
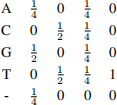
(a) Write down a multiple sequence alignment that corresponds to this profile matrix. [2 marks] (b) How many different alignments are there that correspond to this profile matrix? Are all multiple sequence alignments corresponding to this profile the same size (in terms of number of sequences and alignment length)? Explain your answers. [2 marks]
5. You are given a set of error-free reads s = (AGC, CGC, CTT, GCG, GCT} and you want to reconstruct the DNA sequence X from which the reads originated. Show step-by-step how you will solve this sequence reconstruction problem using the following two approaches:
(a) For the following, use the greedy algorithm described in lectures.
i. Write down the reconstructed sequence and explain how you obtained it. That is, explain the order in which the reads were considered and why you selected that order. [4 marks]
ii. Is the obtained sequence unique? If it is not unique, write down an alternative solution as an ordered sequence of reads. [2 mark]
(b) For the following, use the approach based on the Hamiltonian path. Work with the 2-mer composition of the provided reads.
i. Show the graph and explain how you constructed it. [3 marks]
ii. Explain how you read the sequence from the graph. Provide the reconstructed sequence, if it exists,and explain if the reconstructed sequence is unique. [3 marks]
Section D: Simulation, HMMs and Trees
6. (a) What is the distribution of the random variable that the randomVar pseudo-code below generates given that randomUniform produces a uniformly distributed random variable between 0 and 1? Explain your answer.
randomVar(rate)
var = 0
x = - log(randomUniform())/rate
while x < 1
var = var + 1
x = x - log(randomUniform())/rate
return var
[4 marks]
(b) Given a method PoissPro1(t) that returns the event times of a Poisson process with fixed rate 1 over an arbitrary time period,t, and randomUniform as in part (a), write a pseudo- code method PoissPro(rate, t) that returns the event times of a Poisson process with arbitrary rate over an arbitrary time period, t. Describe one experiment you could run to check the accuracy of your implementation. [4 marks]
7. In the genomes of some phages, the coding regions are richer in purines (bases A and G) than pyrimidines (bases C andT). An HMM with states Y for pyrimindine and U for purine correspond to coding/non-coding regions in these sequences. In the model, the probability that state U is followed by state Y is 3%. The probability that state Y is followed by state U is 1%. The sequence starts in state U 80% of the time.
Each state can emit any of the 4 bases A, C, G, or T. Emission probabilities are given by

(a) Sketch a diagram of the HMM, showing all states, possible transitions and transition proba- bilities. Include the begin state but no end state. [3 marks]
(b) What is the joint probability P (x, π) of the state sequence π = UUUY and the symbol sequence x = ACCC? Leave your answer as a product or sum of numbers. [3 marks]
(c) Suppose you have multiple sequences of the same length x1 , . . . , xn. How could you use P (xj ) for j = 1, . . . , n to decide whether or not the sequences came from the model de- scribed here? [3 marks]
(d) The model was trained using observed sequences. Describe how you could train the algorithm in the case where you knew where coding and non-coding regions were to start with, and in the case you didn’t have this information. What are the strengths and weaknesses of the two training approaches? [7 marks]
(e) Complete the entries a,b, and c in the Viterbi matrix below and draw in the traceback pointers.
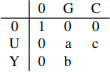
[5 marks]
8. Let the symmetric matrix
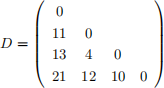
specify the pairwise distances, Dij , between the four sequences x1 , . . . , x4 .
(a) Construct a UPGMA tree from D showing your working. [4 marks]
(b) Perform the first step in the neighbour-joining algorithm on D (up to where the first pair of neighbours i,j are joined and the distances of i and j to the new node are calculated). [4 marks]
(c) Will UPGMA or neighbour-joining (or both or neither) reconstruct the correct tree in this case? Explain your answer. [4 marks]
9. Consider the four aligned sequences, W,X,Y, and Z:
12345
W: TCAAT
X: GCAAG
Y: CGATC
Z: CGATG
(a) Explain parsimony informative means, and identify the parsimony informative sites in the alignment. [3 marks]
(b) By calculating the parsimony score for each possible tree topology for these four taxa, find the maximum parsimony tree. [4 marks]
(c) Add two more columns to this alignment so that the new alignment has a parsimony tree which is topologically different to the parsimony tree for the original alignment. [3 marks]
(d) What typically prevents us from being able to find the maximum parsimony tree and why? [2 marks]
(e) Under what circumstances might we opt to use a parsimony method over a likelihood based method? [2 marks]
10. (a) Describe a heuristic which can be used to try to find the maximum likelihood tree. Will this heuristic typically find the tree that maximises the likelihood? Explain your answer. [4 marks]
(b) Given mutation rate parameter µ and normalised rate matrix Q, how do you calculate the probability that an A mutates to a C along a lineage of length t = 2? (Recall we denote, for example, the (A, A)thentry of a matrix B by BAA .) [3 marks]
(c) Let X and Y be sequences of length L. How can you use the calculation in part (b) to calculate the probability that X mutates into Y over a lineage of length t = 2? Explain any assumptions you are making. [3 marks]
(d) Explain why it is not always possible to estimate the height of a tree and the mutation rate at the same time. What data can be used to overcome this problem? [4 marks]
2023-06-20