BIOC3303 SDLP3 - Mass Spectrometry
Hello, dear friend, you can consult us at any time if you have any questions, add WeChat: daixieit
BIOC3303
SDLP3 - Mass Spectrometry
How to identify and characterize a protein using mass spectrometry
1. Introduction
The aim of this mini-project is to provide students with an insight on how modern mass spectrometry techniques can be used to analyse proteins and determine not only their molecular mass but also information about any modifications that may have taken place and their 3-dimensional structures.
Before starting the course-work, the students should familiarise themselves with the MS lectures (Minerva), on-line tutorials on mass spectrometry and other sources (see Section 4.).
2. Coursework
Q1. Figure 1 shows two electrospray ionisation-mass spectrometry (ESI-MS) m/z spectra of different forms of the protein cytochrome c.
(i) Using all charge states where m/z values are indicated on the spectra in Figure 1, calculate the molecular masses of the two proteins (use the mass of hydrogen = 1.0078 Da). Show your working out.
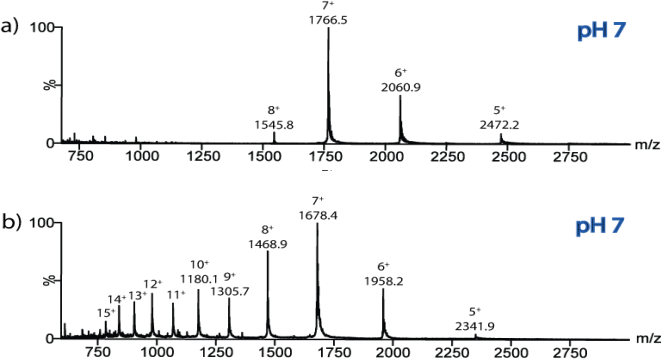
Figure 1. ESI-MS spectra of two differentforms of the protein cytochrome c analysedfrom solution buffered atpH7.
Molecular mass of protein in (a) = (2 marks)
Molecular mass of protein in (b) = (2 marks)
(ii) What is the difference between the two protein masses?
The mass difference is = (1 mark)
|
(iii) |
How could you account for the mass difference between the two protein samples? Draw the structure of the molecular group which accounts for the additional mass, illustrating all atoms involved and the types of bonds it makes with the relevant residues in the protein (3 marks). Give the sum formula of this group (CnHm …) and calculate its mass, taking the bonds formed into account, and check if it matches the experiment (2 marks). Also indicate the position(s) of the residues in the amino acid sequence of the protein where binding occurs (1 mark). How does this binding differ from other proteins in the same family? (give an example; 1 mark). (7 marks) |
(iv) Use an isotope distribution calculator (e.g. http://www.sisweb.com/mstools/isotope.htm)
to analyse the isotopes of a compound with the chemical formula C34H32O4N4Fe. Show the resulting isotope distribution (2 marks) and report the monoisotopic and average mass (2 marks). What is most likely cause for the peaks at 614 and 617 Da? (2 marks). (6 marks)
(v) Describe the differences in the charge state distributions observed in the two spectra (2 marks), and give a possible explanation taking your findings in (iii) into account, and how charging occurs in electrospray ionization (2 marks). (4 marks)
The amino acid sequence of cytochrome c is as follows:
GDVEKGKKIF VQKCAQCHTV EKGGKHKTGP NLHGLFGRKT GQAPGFTYTD ANKNKGITWK EETLMEYLEN PKKYIPGTKM IFAGIKKKTE REDLIAYLKK ATNE
(vi) What is the theoretical molecular mass of this sequence?
You can find this by going to the website:http://www.peptidesynthetics.co.uk/tools/ and pasting the sequence into the mass calculator there.
Theoretical mass = (1 mark)
Is this the same mass that you calculated from either of the spectra in Figure 1? If not, what is the difference in mass and can you suggest an explanation for this (1 mark)? Also comment on the usual biochemical role of the group which corresponds to this mass difference (1 mark). (2 marks)
Q2. The protein whose spectrum is shown in Figure 1(a) was subjected to limited proteolysis for just 15 minutes using the enzyme trypsin.
(i) Why would you use limited proteolysis when investigating protein structure (1 mark), and how does it differ from a full protein digest (1 mark)? (2 marks)
(ii) Where would you expect the enzyme trypsin to cleave a protein (1 mark)? Where does trypsin occur in nature (1 mark)? (2 marks)
A number of peptides were generated by this method and the peptide mixture was analysed using ESI-MS. The m/z values and corresponding charge states for each peptide were measured and are shown in columns 1, 2, and 3 of Table 1.

Table 1. m/z values of the peptides generatedfrom limited trypsin proteolysis of the protein
whose ESI-MS spectrum is shown in Figure 1(a).
(iii) From the m/z values, calculate the molecular masses of each of the peptides (use H = 1.0078 Da) and complete column 4 of Table 1. (6 marks)
(iv) Determine which regions of the protein’s sequence these peptides relate to, and complete column 5 of Table 1 with both the partial sequences and the residue number ranges. (7 marks)
You can deduce this information by going to the website: http://web.expasy.org/peptide_mass/and pasting in the amino acid sequence given above. Then generate the masses of the expected peptide fragments from a tryptic digest of this protein sequence.
(v) From the peptides identified, can you say something about the protein’s 3D structure? There is a lack of peptides detected from the first 30 amino acid residues in the sequence. Show on the protein sequence where you would have expected trypsin to cleave along the protein sequence (2 marks). Can you explain why this N-terminal part of the protein has not been proteolysed, based on what we know now? (2 marks) (4 marks)
GDVEKGKKIF VQKCAQCHTV EKGGKHKTGP NLHGLFGRKT GQAPGFTYTD ANKNKGITWK EETLMEYLEN PKKYIPGTKM IFAGIKKKTE REDLIAYLKK ATNE
(vi) If a full tryptic digest is carried out, into how many non-trivial pieces (i.e. longer than 1
amino acid) is the sequence of the protein cut? Indicate these using the protein sequence (2 marks). Would you expect to detect all these peptides in a proteomics (ESI LC-MS/MS) experiment – and why? (2 marks) (4 marks)
Q3. As further evidence of the 3D structure of the cytochrome c protein whose m/z spectrum is shown in Figure 1(a), the protein was analysed using ESI-travelling wave ion mobility spectrometry (TWIMS)-MS. This technique measures the time (in ms) that it takes the sample ions to traverse a gas-filled drift tube under the influence of an electric field. When interfaced to a mass spectrometer, the m/z values for each ion are measured also, in the same experiment.
When using conventional ion mobility (IM), the cross-sectional areas (Ω) can be measured directly from the drift times; however, when using TWIMS this is not the case. A set of protein standards, whose cross-sectional areas have already been measured elsewhere on a conventional IM-MS instrument [3], must be analysed on the TWIMS-MS instrument, and then used as a calibration for subsequent analyses of “unknown” analytes. This procedure is described in the following tutorial, in Section 3, and also in more detail in [4,5].
Table 2 shows an ESI-TWIMS-MS data set (m/z, z, tD, and Ω) for the ions generated from the protein horse heart apo-myoglobin. The cross-sectional areas for each of the charge states have been taken from measurements made elsewhere on a conventional IM-MS instrument [3].
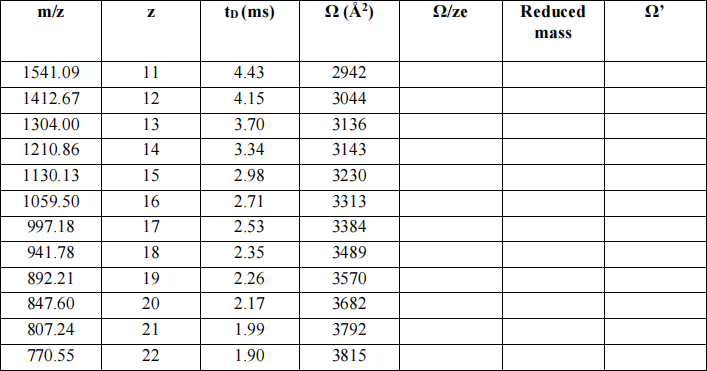
Table 2. IM-MS calibration datafor the protein horse heart apo-myoglobin (mass 16,951.5 Da).
(i) Fill in column 5 in Table 2 (Ω/ze) for each charge state. (e = charge on an electron) (n.b. you may wish to use Excel to work with these data) (2 marks)
The following equation shows the relationship between the mass of the ions (mI) and the mass of the IM buffer gas, in this case nitrogen, (mN, for N2 = 28 Da).
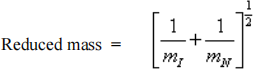
(ii) Calculate the reduced mass for each charge state and complete column 6, Table 2. (2 marks)
The following equation shows the relationship between Ω and Ω’:
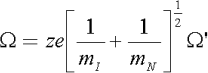
(iii) Now calculate Ω’ for each charge state and complete column 7, Table 2. (6 marks)
(iv) Plot a graph showing Ω’ (x axis) versus drift time (tD) for each of the horse heart apo-myoglobin ions used for the TWIMS calibration. (5 marks)
Using ESI-IM-MS the following drift times (tD) were measured for the holo-cytochrome c 5+ charge state ions:
|
m/z |
z |
tD (ms) |
Ω (Å2) |
Ω/ze |
Reduced mass |
Ω’ |
|
2472.2 |
5 |
2.10 |
2101 914 |
2.6229E+21 1. 1409E+21 |
0.1892 |
6.03E+21 |
|
2472.2 |
5 |
3.75 |
2767 1158 |
3.4537E+21 1.4451E+21 |
0.1892 |
7.94E+21 |
Table 3. IM-MS protein datafor holo-cytochrome c (m/z, z, tD).
(v) Using the molecular mass you calculated in Question 1, complete the “Reduced mass” column in Table 3 (column 6). (1 mark)
|
(vi) |
From the calibration graph you have drawn, use the drift times (tD) measured for the two 5+ charge states ions of cytochrome c given in Table 3 to find the corresponding Ω’ values and enter these into column 7, Table 3. (2 marks) |
|
(vii) |
Using the equation above and the values of Ω’ and the reduced mass, calculate Ω/ze and finally Ω for the two 5+ charge state ions and complete columns 5 and 4, respectively, in Table 3. (2+2 marks) |
(viii) Can you suggest why there are two 5+ charge state ions, of the same molecular mass and
m/z? what does this tell us about the protein when analysed under these particular conditions? (3 marks)
Q4. Describe how hydrogen-deuterium exchange (HDX) MS can be used to characterize epitopes and paratopes of antibody/antigen binding. Describe the principle of the experiment and how it is carried out, including all the key steps, and how the data is analysed and what information you gain.
Max. 250 words (excluding references), you can include 1 figure (25 marks)
3. Tutorial: Calculating collision cross-sectional areas using ESI-IM-MS.
Conventional IMS measures the time it takes an ion to migrate through a buffer gas in the presence of a uniform electric field.
K = mobility of ion; vD = the drift velocity (vD) of the ion; E =electric field. The drift velocity (vD) = drift time (tD) to traverse the length (L) of the drift cell (Equation 1):
(Equation 1) 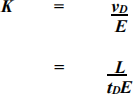
The mobility of an ion depends on the buffer gas density, so the reduced mobility (Ko) is generally reported (Equation 2):
(Equation 2) 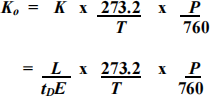
where P = buffer gas pressure and T = buffer gas temperature .
With conventional IM, the mobility of an ion (K) can be calculated directly from its observed drift time, and from this the collision cross-sectional area (Q) can be calculated (Equation 3):
(Equation 3)
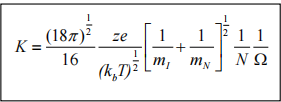
where, z = number of charges on the analyte ion,
e is the charge on an electron (1.6022 x 10- 19 C),
kb is the Boltzmann constant,
T is the temperature,
N is the number density of the buffer gas,
and
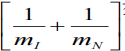
is the reciprocal of the reduced mass between the analyte ion (mI) and the buffer gas of molecular mass mN.
The proportionality between the collision cross-section (in Å2), and the average drift time (tD) of a given ion of molecular mass mI and nominal charge z, in which P is the buffer gas pressure (in torr), T is the temperature (in K), and L the length of the drift cell is given by (Equation 4):
(Equation 4)
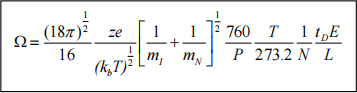
The primary difference between conventional IM and the TWIMS system is that the electric field is not constant and uniform; the voltage pulse across the series of stacked rings creates a travelling wave of electric potential that varies with both time and the length of the drift tube. Derivation of a relationship between Q and tD for the TWIMS system is more complex.
Therefore the drift times of ions of known collision c
2023-03-20
How to identify and characterize a protein using mass spectrometry