CHEN E4880 Atomistic Simulations Properties of an Elemental Transition Metal
Hello, dear friend, you can consult us at any time if you have any questions, add WeChat: daixieit
CHEN E4880 – Atomistic Simulations – 2023/02/02
Project 1: Properties of an Elemental Transition Metal
(Due on February 26, 2023 at 11:59 PM)
Transition metals are of great technological relevance for the chemical industry due to their unique properties and versatility in various applications, such as catalysts, magnetic materials, and electronics. In this project, you will explore the properties of an elemental transition metal, for now, in the absence of temperature or pressure.
A technical objective of this project is to become familiar with the LAMMPS software and its input/output file formats. Scientific objectives are to find the ground-state energy, lattice parameter, vacancy formation energy, and one surface energy of face-centered cubic (FCC) Pt. As you work through the project, you will also learn about the importance of convergence parameters and the limitations of interatomic potentials used in atomic-scale simulations.
Geometry optimizations (relaxations)
In nature, a crystal in equilibrium is automatically in the lowest energy (ground-state) configuration. The ground-state lattice parameters and atomic positions define the ground-state lattice geometry that minimizes the lattice energy. The ground-state lattice geometry is generally not known a priori. The best we can do is to start with a reasonable guess and try to find the lowest energy configuration from there. In geometry optimization or relaxation the atomic positions and lattice parameter(s) are adjusted until an energy minimum is found.
Vacancy formation energy
The vacancy formation energy Ev is defined as the energetic cost to remove an atom from a lattice site and reinsert it into the bulk of the material:
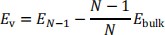
where Ebulk and E"#$ are the energies of the perfect bulk supercell with N atoms and of the defect cell with one atom less, respectively. Ebulk is proportional to the number of atoms in the supercell, so we can scale it with factor (n − 1)/n in order to compare the energy of a defective cell with n − 1 atoms to that of a perfect cell with n – 1 atoms.
Surface energy
The surface energy is the energy that is required to truncate an infinitely extended crystal along a specific lattice plane. Calculating surface energies follows a similar overall approach to calculating vacancy formation energies, but different convergence parameters must be considered.
Surface relaxations and pair potentials
The Lennard-Jones potential is given by the following equation
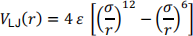
Pair potentials, such as the Lennard-Jones potential, show outward surface relaxations, which is in disagreement with experimental observation.
1. Lattice constant of Pt
a. Calculate the lattice constant (in Å) and total energy (in eV) for FCC Pt using the supplied LJ potential. First, use LAMMPS’ built-in minimizer (input file lmp-in.1a-relax), then find the lattice constant by manually adjusting the lattice parameter (input file lmp-in.1a-single). Start with a lattice parameter close to the relaxed lattice parameter. Plot the energy as a function of the lattice parameter from above to below the optimized value (Figure 1), and identify the equilibrium lattice constant.
Hint: See the project guide for an explanation of the LAMMPS input format.
b. Repeat the calculations, both automated and manual (Figure 2), for the supplied embedded-
atom model (EAM) potential (input file: lmp-in .1b-single).
Hint: Don’t forget to upload the EAM potential file to nanoHUB.
c. Discuss your findings. The experimental lattice constant of Pt is 3.92 A. How do the calculated lattice constants compare to the experimental value? Is this expected? Is it significant that the
absolute values of the lattice energies for the LJ and EAM potential are different? Hint: Consider how the different potentials were constructed (see table below).
2. Vacancy formation energy of Pt
a. Compute the vacancy formation energy (in eV) with the provided LJ potential as a function of the supercell size. Do not relax the atomic positions after taking out an atom. Perform a convergence test and plot the energy against the convergence parameter (Figure 3). What is the ratio of the vacancy formation energy to the cohesive energy? Explain your result. Explain how you calculated the cohesive energy. Hints: The (conventional) FCC unit cell contains 4 atoms. Use the optimized LJ lattice parameter from problem 1.
b. Repeat your calculation, but this time relax the atomic positions after creating the vacancy. Perform another convergence test and add results to Figure 3. How does the vacancy formation energy change compared to the unrelaxed calculations? What is the reason for this behavior? Hint: Adjust your LAMMPS input file for relaxations (see problem 1).
c. Now compute the vacancy formation energy using the provided EAM potential. Do it as accurately as you can. Report your convergence test (Figure 4). Use what you learned in parts a. and b. Hint: Use the optimized EAM lattice parameter from problem 1 as initial value.
d. The experimental vacancy formation energy is 1.6– 1.77 eV. How do your final results compare? Discuss your result in light of your findings from problem 1.
3. Surface energy of the Pt(100) facet
a. Compute the surface energy of the Pt(100) surface using the LJ potential. Report all equations. What are the two convergence parameters? Perform and plot your convergence tests (Figure 5). Report your result in meV/Ǻ2 . Be computationally efficient.
b. Repeat with the EAM potential, including convergence tests (Figure 6). Please skip relaxations for problem 3.
4. Short answers
a. For what class of compounds are Lennard-Jones potentials most suitable (name 1 example)?
b. For what class of compounds/materials are EAM potentials most suitable (name 1 example)?
c. Name 2 examples of materials/interactions for which neither LJ nor EAM are appropriate.
5. Conceptual understanding
a. Calculate the distance r+ where the LJ potential reaches its minimum. Express r+ in terms of e and ![]() , and evaluate using the values from your calculations (e.g., lmp-in.1a-single).
, and evaluate using the values from your calculations (e.g., lmp-in.1a-single).
b. Determine the nearest-neighbor distance dNN in the optimized structure of problem 1. Why is dNN different from r+ ?
c. Based on your results, explain why computer simulations are required even for simple energy models such as the LJ potential.
Assignment
1. Address the above questions in a two-page report.
a. Introduce subsections to distinguish between the different problems.
2. Include Figures 1–6 in the appendix. Make sure to refer to the figures from your report.
3. Include all derivations (problem 5) in the appendix. Pictures of hand-written notes are fine.
4. Include examples of input files for problems 1–3 in the appendix.
Provided Files

The Lennard-Jones potential was constructed as a loose fit to the lattice constant and the vacancy formation energy.
Parameters: ε = 0.200 eV; σ = 2.540 Å
The EAM potential was fitted to sublimation energies, elastic constants, and vacancy formation energies.
Problem 2 and 3:
We do not provide separate files for problem 2 and 3. Use the files provided for problem 1 and start from there. See the project guide for hints regarding the creation of vacancies and surface slab models.
2023-02-17